Hyperkalaemia And Hypokalaemia
Overview
The normal plasma concentration of K+ is 3.5-5 mmol. Most (98%) of the K+ in the body is sequestered intracellularly, particularly in skeletal muscle. Any process that allows K+ to leak from cells on a large scale will lead to hyperkalaemia. The homeostatic balance of dietary K+ intake and renal excretion of K+ is crucial to maintaining K+ in the normal range. The concentration of K+ in plasma is critical because K+ is a crucial determinant of membrane potential in excitable cells. Increased K+ depolarises such cells. The heart is particularly sensitive to alterations in K+, and these will probably show up as changes in the ECG before effects in other muscles are obvious. When cardiac myocytes are depolarised, an increased number of Na+ channels are inactivated, hence cardiac excitability is decreased in hyperkalaemia (see this review). Hyperkalaemia will eventually lead to cardiac arrhythmia and sudden death if allowed to worsen unmanaged.
Hormonal regulation of potassium homeostasis
Probably the most influential mediators of K+ homeostasis are the catecholamines noradrenaline and adrenaline. Activation of β2-adrenoceptors by agonists such as salbutamol can lead to hypokalaemia. There’s also an opposing effect by α-adrenoceptors leading to hyperkalaemia. As such, administration of a mixed adrenoceptor agonists such as adrenaline can have mixed effects, depending on the dose and the (patho)physiological context. In principle, β2-agonists could be used to treat hyperkalaemia, but the side-effects at the doses required to produce such an effect (tachycardia, muscle tremors) out-weigh any perceived benefits. Unsurprisingly, the regular use of β2-agonists as bronchodilators in the treatment of asthma is associated with mild hypokalaemia.
The discovery of insulin and its subsequent use by diabetics lead to the serendipitous discovery that this hormone also regulates potassium homeostasis. Insulin increases the activity of Na+/ K+-ATPase pumps, leading to pumping of K+ into cells, an effect that is exploited therapeutically in hyperkalaemia. Of course, insulin more famously lowers blood glucose, so when it is used to treat hyperkalaemia supplemental glucose is given at the same time to prevent hypoglycaemia. On the other hand, it follows that when insulin is given to lower blood glucose (as in diabetic ketoacidosis), supplementary K+ is often required.
Common causes of hyperkalaemia
Pseudo-hyperkalaemia:- Haemolysis
- Thrombocythaemia
- Ischaemia e.g. tourniquet
True Hyperkalaemia:
- Rhabdomyolysis and burns
- Oliguric renal failure
- Some drugs (K+-sparing diuretics, ACE inhibitors, suxamethonium)
- Addinson’s disease
- Metabolic acidosis
Pseudo-hyperkalaemia
A blood sample may falsely suggest hyperkalaemia for a couple of reasons. Firstly, if there is a significant delay between taking a blood sample and analysing it, some haemolysis (rupture of red blood cells) may occur resulting in the release of their intracellular K+. Secondly, in thrombocythaemia (excess platelet counts) the increased number of platelets in a blood sample will release more K+ than is usual when it clots. Finally, the use of a tourniquet when taking a blood sample can lead to peripheral ischaemia and some cell death, also leading to K+ release. Another theory to explain this phenomenon suggests that excessive fist clenching to pump blood into veins prior to venous blood sampling leads to K+ release from working muscle cells (due to the K+ efflux associated with the repolarisation phase of the muscle action potential).
Hyperkalaemia
Rhabdomyolysis and burnsBecause 98% of the body’s K+ is located intracellularly, any process that leads to widespread cell death will lead to hyperkalaemia. The breakdown of damaged skeletal muscle (rhabdomyolysis) and large scale burns are two of the commonest such causes.
Oliguric renal failureSince K+ is excreted primarily by the kidneys (there is a small component excreted in faeces), failure of the kidneys to produce urine is an obvious cause of the accumulation of K+ and hence hyperkalaemia.
Drug-induced hyperkalaemia and Addison’s diseaseSome drugs also produce hyperkalaemia by affecting K+ homeostasis. Diuretics that increase K+ reabsorption by the kidney (so called potassium-sparing diuretics such as amiloride) will clearly have this effect. Angiotensin converting enzyme (ACE) inhibitors also increase plasma K+, although this is a little less straight forwards. ACE inhibitors prevent the conversion of angiotensin I to the more active peptide angiotensin II. One of the effects of angiotensin II is to cause the secretion of aldosterone by the adrenal gland. Aldosterone increases K+ secretion by the kidney. So by inhibiting angiotensin II production, and hence aldosterone release, potassium is retained by the body leading to hyperkalaemia. Similarly, in Addison’s disease in which the function of the adrenal gland is compromised, aldosterone production is inhibited and hyperkalaemia can result. Finally depolarising neuromuscular blocking agents (used in producing paralysis) such as suxamethonium can cause mild hyperkalaemia. This may be because during the constant depolarisation of muscle fibres that they produce muscle cells attempt to repolarise by opening K+, which then leaks into the extracellular compartment. This mild effect (an increase of around 0.5 mmol) is generally tolerated or compensated for but could worsen another trigger for hyperkalaemia. For example, consider a patient with extensive burns (see above) who is put into a medically-induced coma (including suxamethonium) and mechanically ventilated.
AcidosisThe effects of acid base disturbances on K+ redistribution are clear and have been for many decades. However, finding a good explanation of how this occurs can be hard work. Many basic physiology texts omit any discussion of this phenomenon and many clinical texts simply take it as read: in acidosis, H+ goes into cells and K+ leaks out. Searching the internet reveals various theories and pronouncements with no clear consensus. This background should ring alarm bells for students: when it’s hard to find an answer, we probably don’t really know.
What we do know is that muscle cells, which make the greatest contribution of K+ elevation, do not possess a single protein that we could call a H+/K+ exchanger. We also know that isolated muscle in a test tube will release K+ in the solution surrounding if it is made acidic, and that the cells appear to sequester H+ in the process. So, K+ is exchanged for H+ somehow, but it’s not due to a single process. There are so many pumps and exchangers in muscle cells, almost any “just-so” story can be constructed, but the following below are probably the most important mechanisms by which this occurs:
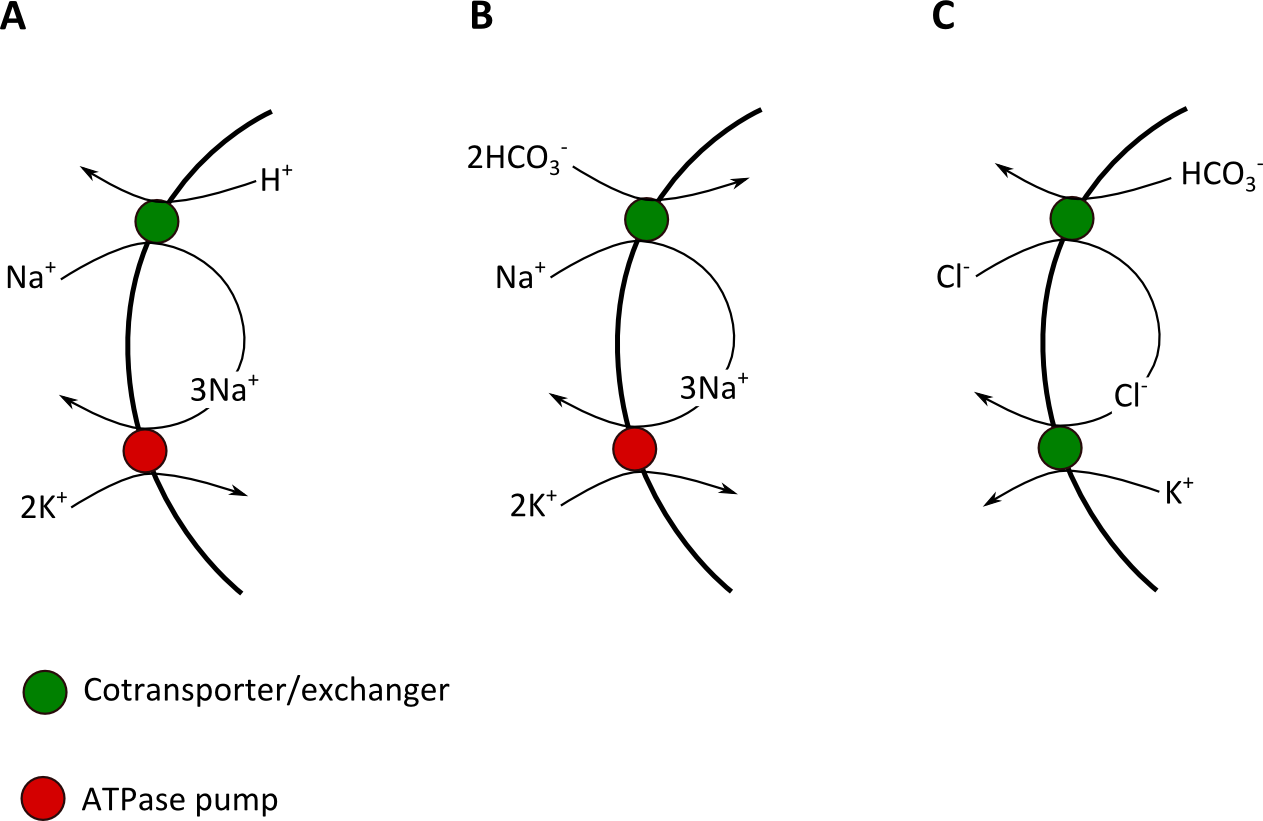
Metabolic acidosis tends to cause a more significant hyperkalaemia than acute respiratory acidosis, possibly in part because in uncompensated respiratory acidosis plasma HCO3- is normal rather than decreased, so the mechanisms in Figure 1 B &C contribute much less (if at all) to the increase in plasma K+.
Renal effects of acidosis
Renal K+ regulation is complex. Again, it can be difficult to find a consistent textbook version of events to explain why acidosis causes retention of K+ by the kidney such are the many pumps, cotransporter/antiporters, ion channels and different cell types along the length of a nephron. It’s probably simpler to consider the kidney as a whole and how acidosis affects overall K+ than to examine the effects of increased H+ on individual proteins responsible for movements of ions at various points along the nephron. As with the systemic effects of acidosis on K+ redistribution (see above), no single transport system is responsible for increased renal retention of K+ in response to changes in H+ and HCO3-. The overall acute effect of acidosis is increased secretion of Na+ into the urine and increased K+ reabsorbed into the plasma leading to hyperkalaemia.
Common causes of hypokalaemia
The causes of hypokalaemia are also many and varied, but make sense physiologically once you come to grips with them:
- Diuretics
- Vomiting and diarrhoea
- Cushing’s syndrome and similar
- Conn’s syndrome
- Alkalosis
- Renal tubular failure (e.g. recovery from acute kidney injury)
Hypokalaemia
DiureticsHow can diuretics cause both hyperkalaemia (see above) and hypokalaemia? It depends on the drug and how it works. Potassium-sparing diuretics do just what their name suggests: while some ions and fluid are lost, potassium is retained. These diuretics can cause hyperkalaemia. Other diuretics (especially loop diuretics – e.g. furosemide - and thiazides – e.g. hydrochlorothiazide) act on different ion pumps and transporter and can lead to potassium loss and hypokalaemia.
Vomiting and diarrhoeaVomit doesn’t contain much K+, but it does contain a lot of H+, which has to be replaced from the plasma. So, persistent vomiting can lead to metabolic alkalosis. This produces the opposite effect as acidosis: K+ is pushed into cells in exchange for H+. There’s a further possible complication too: if fluid loss from persistent vomiting is significant, increased aldosterone secretion by the adrenal gland will decrease K+ reabsorption by the kidney. So, persistent vomiting can cause hypokalaemia not only by producing metabolic alkalosis, but also by reducing blood volume.
Diarrhoea on the other hand leads to inadequate colonic reabsorption of HCO3- which produces metabolic acidosis. You might expect this to cause hyperkalaemia, since that is the general effect of metabolic acidosis (see above). However, although it is a minor component of K+ homeostasis (compared to renal mechanisms), some K+ is lost in the faeces, and much more is lost in diarrhoea than usual. So, in this setting you generally see the unusual combination of hypokalaemia with metabolic acidosis.
Cushing’s syndrome and similar
In Cushing’s syndrome levels of endogenous cortisol or (more usually) exogenous synthetic glucocorticoids are high. While cortisol usually bind to glucocorticoid receptors (synthetic glucocorticoids are quite selective for this receptor), they can also bind to the receptor for aldosterone, the mineralocorticoid receptor (MR). This usually doesn’t occur, because an enzyme (11β-hydroxysteroid dehydrogenase) in the kidney breaks cortisol down, preventing it from acting. However, a high enough plasma cortisol concentration can overcome this control system and cortisol binds to the MR mimicking aldosterone and causing retention of Na+ at the cost of a net loss of K+ (see above). Synthetic glucocorticoids tend not to cause this problem as they have been developed to have very low affinity for the MR.
Conn’s syndromeIn Conn’s syndrome an adrenal adenoma releases excessive aldosterone (hyperaldosteronism), causing renal Na+ retention and K+ loss. Non-Conn’s hyperaldosteronism will have similar effect.
AlkalosisThe effect of alkalosis on K+ regulation is the opposite of the effect of acidosis (see above). This includes both the systemic and renal effects. Now, that’s an easy one to remember.
Renal tubular failure (e.g. recovery from acute kidney injury)Although acute kidney injury will initially lead to hyperkalaemia due to failure to reabsorb K+, if the injury results in renal tubular necrosis then the damage will take some time (1-3 weeks) to heal. It is not unusual for glomerular filtration to recover before the tubules have restored function, and during this recovery phase urinary output will increase while tubular ion reabsorption will be dysfunctional. This leads to urinary wasting of K+ and hypokalaemia.