Blood Coagulation
Blood coagulation
Getting your head around the coagulation cascade isn’t easy. There are multiple connections in the system and each version that you look at seems to be different. In some cases they are merely drawn differently, in others the information is out of date or poorly put together. The system is so complex, that any attempt to make a simple summary is necessarily a set of convenient omissions and lies. Everyone makes their own omissions and weaves their own lies to help make sense of the system. Add to that the problem that the coagulation factors are numbered relative to their historical discovery, rather than in the order that they relate to each other and you have the potential for serious confusion. There are two basic models of the coagulation cascade which have their pros and cons.
The first is the standard “map” of the coagulation cascade showing how the different factors go together. Everyone has tried to make the “perfect” map that shows everything, but really they’ve made the map that works best for them. The map we’ll run through below should have broad appeal because it’s a synthesis of several attempts to communicate this information clearly. Hopefully, it will work for you. This old map is an artificial system based on older ideas about coagulation and different clotting tests, but it still has relevance today, just as the same tests do. Furthermore you can cram a lot of useful information into it.
The second way to look at blood coagulation (coming soon) is the cell-based model. This shows more clearly how the vessel wall, platelets and coagulation factors interact during formation of a clot, but you can’t squeeze as much detail into it without making a complete mess. There’s nothing as frustrating as finally figuring out how something works then returning to it later when revising and finding that it’s all turned to spaghetti in your mind again because your summary is a mess.
What’s the best way to get your head around the mechanics of blood coagulation? Probably a mixture of the two approaches. If you only know the cell-mediated model, you’ll struggle to understand laboratory coagulation testing. If you don’t understand how the cell-mediated approach shows how blood clots in vivo, then you’ll struggle to understand how blood disorders and drugs can cause imbalances in the system.
What you will need:
- A piece of paper – landscape orientation is easiest
- Some acetates/transparencies/overhead projector sheets for overlays
- Different coloured felt-tip pens
Start where the action is
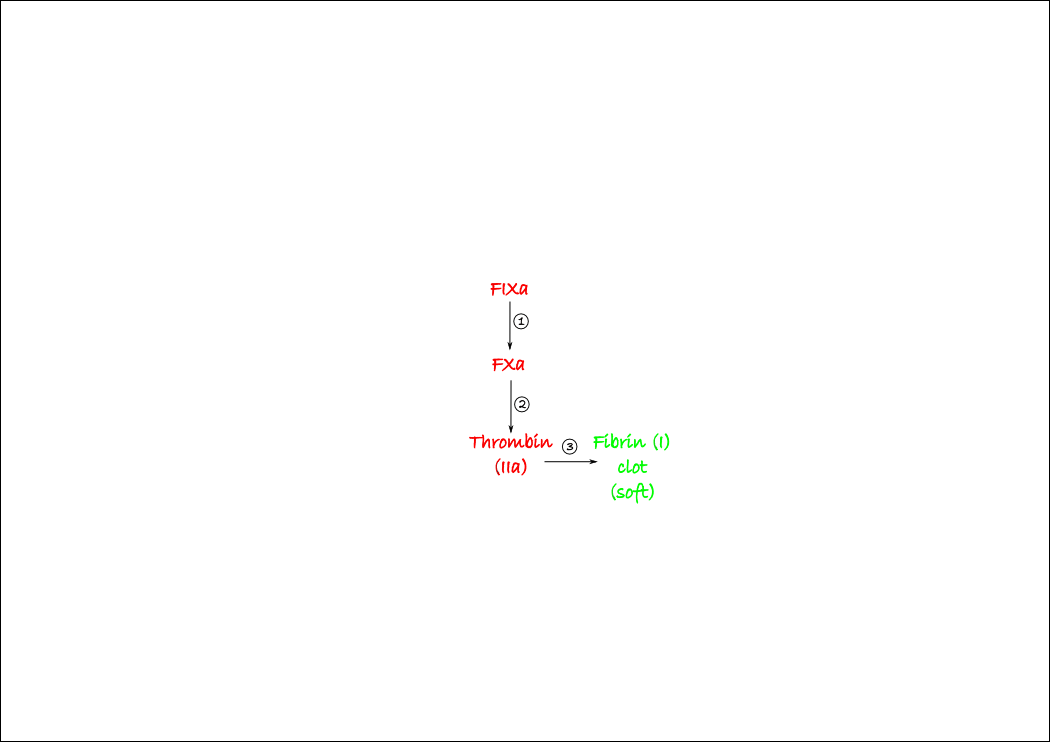
Let’s begin with thrombin, as it is the main player in coagulation. Thrombin (AKA FIIa) is a protease that cleaves fibrinogen into fibrin. Fibrinogen is a big long molecule that is dissolved in plasma. Once it is cleaved it becomes insoluble fibrin and forms the basis of the clot gel. Thrombin is generated by the cleavage of prothrombin by another protease, Factor Xa (FXa). Similarly, FXa is activated from FX by FIXa. You can draw this very simply near the middle of your page. Number each step in their order; this is going to be really helpful later on when we add the other components of the cascade. Note that we’re only showing the activated factors – this simplifies things enormously. All you need to remember is that any activated factor (e.g. FXa) has an inactive precursor (e.g. FX).
Thrombin activates two important cofactors, FVa and FVIIIa. Neither is a protease, but both require cleavage by thrombin to become activated. Using Ca2+ ions in plasma, these cofactors help bring together two complexes:
- The tenase complex: FVIIIa+FIXa to rapidly generate FXa
- The prothrombinase complex: FVa+FXa to rapidly generate active thrombin
These complexes form on charged surfaces. In a laboratoy setting, this would be glass. In the body, activated platelets express charged phosopholipids (PL on the diagram below) in their cell membranes, and the complexes form on these. You can represent this on your diagram using different colours like this:

The thicker blue lines represent the accelerated nature of the process and the fact that cofactors are required for this to occur (you’ll see the utility of this soon).
Once enough thrombin has been generated, FXIII is activated, which crosslinks the fibrin fibres into a solid clot. FXIIIa is not a protease (it’s a transglutaminase, for what it’s worth) so make it a different colour. Once you’ve added this to your diagram, you have the entire (well, as much as a normal person needs) common clotting pathway figured out. This is the bulk of the coagulation cascade so we’re almost done.

We’re now left with addressing the problem of how the common coagulation pathway gets activated in the first place. Two pathways are traditionally identified on the basis of how blood can be made to clot in a laboratory. The early history of our understanding of clotting came from such laboratory work, which is why it has left an impression in the way we draw the clotting cascade. We still use clotting tests based on this distinction, so it is a useful one, but it is necessarily artificial.
The most important initiator of clotting is Tissue Factor (TF). This is known as the extrinsic pathway because you need to add something (TF) to blood to get it to clot. TF is a receptor for FVIIa, another protease coagulation factor. FVIIa binds much better to TF if it is bound to a negatively charged surface of phospholipids with calcium, in the same way that FIXa and FXa work more efficiently when similarly bound with their cofactors (the bits in blue we’ve drawn already). Again, activated platelets provide this surface. Once activated, VIIa activates FXa and the common pathway is initiated. You can add this to your diagram, but a better strategy would be to put a transparency over the top and continue on that. In this way, when you come back to study this again, you can begin again with just the common pathway and add the rest piece by piece as you go.
![]()
Where does TF come from? It’s not usually expressed by endothelial cells (but may be in inflammatory conditions), but is present on most subendothelial cells. So if the endothelium is damaged – as it will be if a vessel is cut or broken – TF is exposed, ready for FVIIa to bind to and start coagulation. This slow process will be accelerated at several key points (the blue ones) by the presence of activated platelets.
The other activation pathway is the intrinsic or contact pathway. This is why blood clots in a glass tube or if exposed to collagen – as it would be in a damaged vessel since collagen makes up the connective tissues under the endothelium. How glass or collagen lead to activation of FXII represents our first substantial cheat. Look it up by all means, but we’re not going to need it to understand bleeding disorders or the effects of drugs. Without requiring cofactors or platelets, FXII activates FXIa, which goes on to initiate the common pathway at FIXa. If we add these two steps, we have all the handy bits of the coagulation cascade mechanistically linked together:

Heparin and antithrombin
Now we can make some more transparencies to cover the specifics of how drugs and bleeding disorders interfere with the system. Let’s start with antithrombin. Antithrombin is a protein made by the body which acts as a “suicidal” protease inhibitor. It becomes irreversibly bound to a protease in the process of inhibiting it, so both proteins are taken out of action. This means that either antithrombin or the protease could become depleted in the process. Despite its name, antithrombin inhibits several other proteases of the coagulation cascade with varying efficacy. Make an antithrombin layer with a new transparency and cross out the factors inhibited by antithrombin. Big crosses represent where antithrombin has the greater effect:

Although you can give antithrombin to patients, we very often just activate the antithrombin that is already present in the body. This is what heparin does. Heparin mimics a similar, endogenous molecule (heparan) which provides some structural support to antithrombin, holding it in the optimal three-dimensional configuration. Endothelial cells have heparan molecules bound to their surface, providing just one more mechanism to prevent inappropriate coagulation. Without heparan or heparin the activity of antithrombin at inhibiting proteases is several thousand times less. Because anthithrombin is present in normal blood, you can prevent blood from clotting in a test tube just by adding some heparin. Some blood collection tubes come pre-prepared with heparin in them.
Low molecular weight heparin (shorter chains) when combined with antithrombin form a complex that is more selective at inhibiting FXa. This has been used to more delicately tinker with anti-coagulation than the brute force of high molecular weight heparins which inhibit many coagulation factors. Synthetic heparin-like molecules such as fondaparinux work in much the same way.
Antithrombin represents a natural brake on coagulation that we can trigger with heparin, but there are other such brakes at play which we can’t exploit so easily. That’s not to say that they are not important, because without them coagulation runs out of control. Limiting coagulation is constantly going on in the body on a small scale. Deficiency in these inhibitors (e.g. mutation) leads to increased risk of thrombosis.
Direct FXa inhibitors
More recently, drugs which selectively and directly (i.e. not requiring antithrombin) inhibit the proteolytic activity of FXa have been developed. These have many advantages over older therapeutics, including oral availability, more rapid onset of action and fewer drug interactions. Drugs of this class include rivaroxaban and apixaban.
Direct thrombin inhibitors
Drugs that selectively inhibit the activity of thrombin are also now available and include argatroban and dabigatran. These drugs do not currently have broad applications and are generally prescribed with other therapeutics are contraindicated.
Activated Protein C
One quite important inhibitor of coagulation is activated protein C (APC). APC is a protease that is activated by thrombin (via some complications that we’ll ignore here) and cleaves the amplifying factors FVa and FVIIIa into inactive forms. This represents a negative feedback loop that must be overcome for coagulation to proceed. Coagulation is starting and stopping all the time. We can make a simple additional transparency to show how and where APC acts.
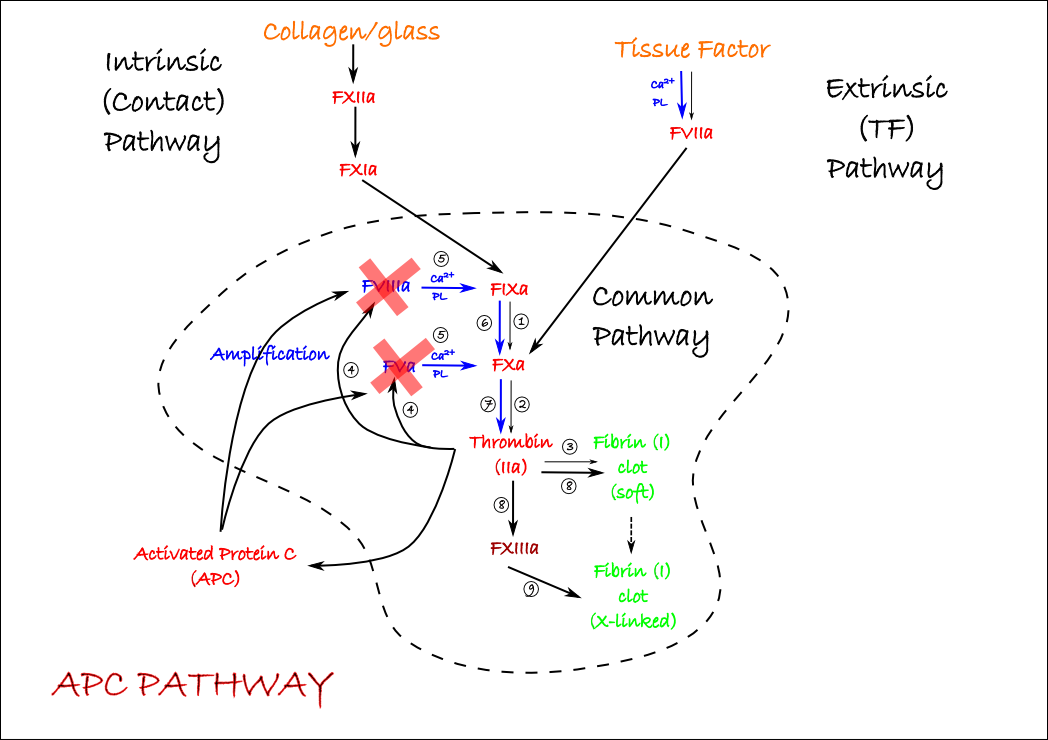
Tissue Factor Pathway Inhibitor
The last important endogenous inhibitor of coagulation is Tissue Factor Pathway Inhibitor (TFPI). This short peptide requires no activation, inhibits FVIIa, and once more represents a natural braking system which must be overcome for coagulation to proceed. We can stick both the APC and TFPI pathways on the one transparency to keep them together.

Warfarin and Ca2+ chelators
We can make another transparency to show how warfarin and related drugs work. Warfarin is a drug which was discovered after a farmer noticed that his cows had severe bleeding disorders after eating certain clover plants. The active molecule was isolated and optimised to make warfarin at the Wisconsin Alumi Research Foundation (the WARF in warfarin). Those protease coagulation factors that require a negatively-charged lipid surface and Ca2+ to work more efficiently have a domain in their structure which contains several carboxyglutamate (or GLA) residues. These are added after the protein is synthesised by an enzyme that requires vitamin K as a cofactor. Warfarin interferes with recycling of “used” vitamin K, effectively causing its depletion in cells, so that these clotting factors are released into blood without this crucial modification. This prevents all the amplification steps in the cascade where these proteases are present – all the blue bits. A deficiency in dietary vitamin K will have the same effect, and administering excess vitamin K is an antidote to warfarin.

Remember, these proteases can still function (thin, black arrows), just not at their accelerated rate because they can’t bind to activate platelet phospholipids and/or combine with cofactors. Also, because warfarin inhibits post-translational modification of these proteases inside cells, it can only work in the body. It’s no good as an anti-clotting agent if added to a blood sample in a test tube, although a blood sample from a patient treated with warfarin won’t clot as readily on glass. Because activation of APC is also a vitamin K dependent process, the initial response to warfarin therapy may be increased coagulation through the loss of this endogenous inhibitor. This reflects the different rates of turnover of each of the coagulation/anti-coagulation factors.
All of the parts of the pathway inhibited by warfarin also require Ca2+. In vitro, we can produce a similar effect to warfarin by removing calcium from blood with a Ca2+ chelator. These are molecules that form complexes with Ca2+ and effectively mop them up. Sodium citrate or other chelators are added to blood sampling tubes to inhibit coagulation when required. You wouldn't use a chelator in a patient, as that would mop up plasma Ca2+ and arrest the heart.
Fibrinolysis
The body has its own way of clearing up a clot: the fibrinolytic system. This simply consists of a precursor protease (plasminogen) which is cleaved into the active form (plasmin) by any protease which has plasminogen activating efficacy. The body’s activators are tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (uPA, or more simply these days, urokinase). Factor XIIa can also activate plasminogen. Once activated, plasmin attacks the fibrin clots, producing fibrin degradation products, including D-dimers which are used in clinical tests to assess coagulation status. One final transparency concludes this simplistic tour of the coagulation system:
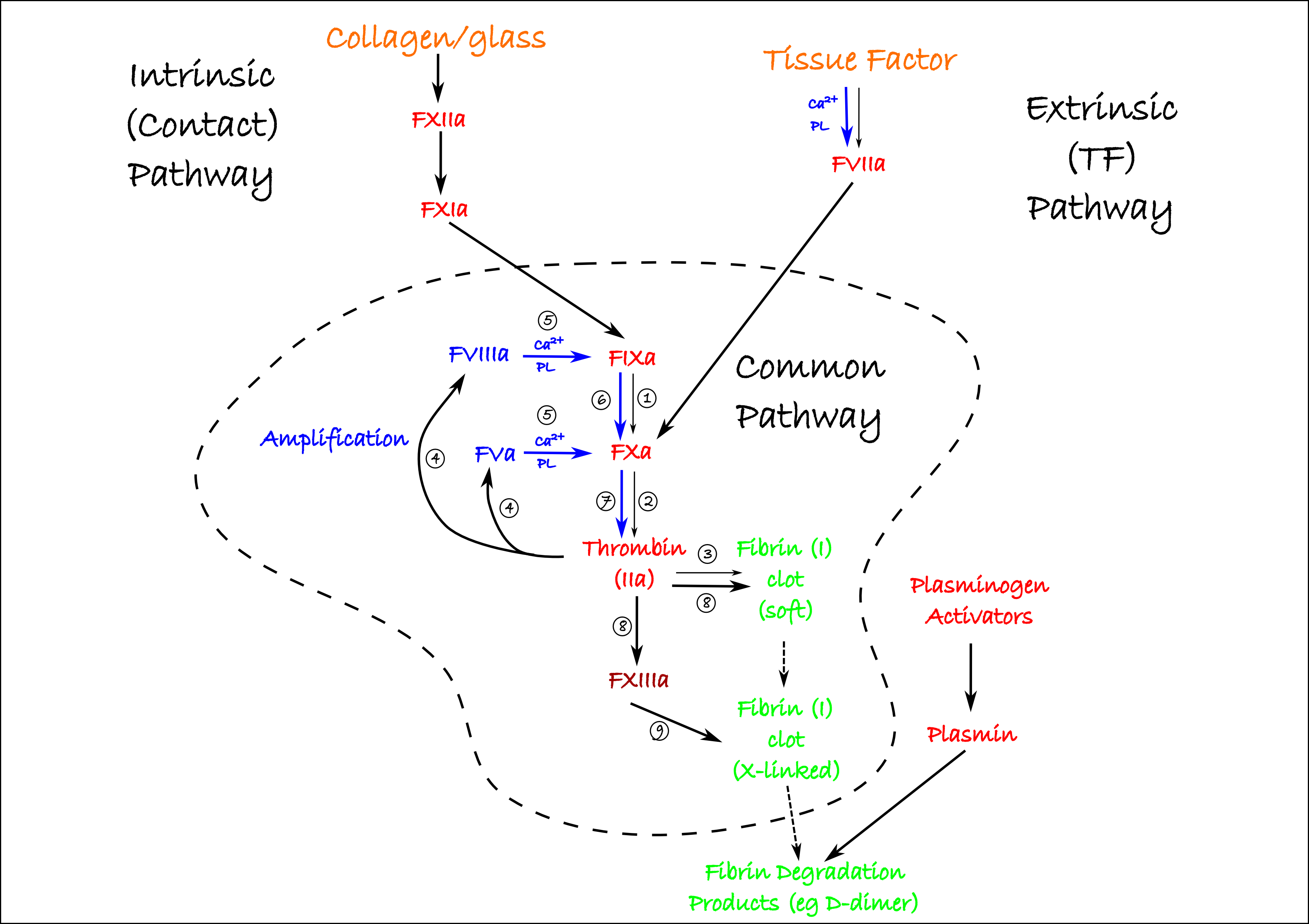
Clinically, streptokinase has been used to activate plasmin. This enzyme is isolated from Streptococcal bacteria which appear to use it to evade getting trapped in blood clots. Being a foreign protein, antibodies may be produced after streptokinase has been administered. These may inactivate the enzyme, or cause anaphylaxis so streptokinase is not used more than once per patient. A way around this problem is to use recombinant tPA such as tenecteplase which are based on the human tPA structure and far less immunogenic. However, they are considerably more expensive.
Haemophilia
There types of haemophilia that comprise the majority of cases: A and B. Haemophilia A involves a mutation of the accelerating cofactor VIII and represents about 80% of cases. The less common (about 20%) haemophilia B involves a mutation of FIX. We can mark these two on another transparency:

Either of these mutations has a serious effect on the clotting cascade, as you can see once you mark them on your map. The severity of haemophilia varies according to how inactive the mutation makes the coagulation factor concerned. For example, there are hundreds of different mutations which can produce defective FIII (haemophila A), each of which has a slightly different effect on the function of the factor. The simplest form of treatment for haemophilia is administration of active factors separated from normal plasma (cheaper) or produced recombinantly (expensive).